
Developing drug combinations is a complex process with challenges and opportunities that can significantly impact patient outcomes. Diseases such as cancer often involve multiple molecular pathways, so single drugs may not be effective enough. This has led to a growing interest in combination therapies that can target different aspects of the disease simultaneously. However, developing these combinations is difficult due to scientific, economic, regulatory and logistical hurdles.
We explored these challenges and strategies for overcoming them in a recent panel discussion, Innovative Therapeutics: Challenges and Opportunities in Developing Drug Combinations, followed by a Q&A. Here are some audience questions we received, answered by Clarivate consultants:
Q: How are novel combinations regulated in different regions, i.e. when neither of the two medical components (non-medical device) is authorized?
In the U.S., the Food and Drug Administration (FDA) evaluates combination products (CP) under the Office of Combination Products (OCP). If neither component is authorized, the FDA typically requires premarket approval (PMA) or a new drug application (NDA) for the combination. The specific pathway depends on the primary mode of action of the combination. The FDA may also require extensive clinical trials to demonstrate safety and efficacy.
In the E.U., the regulation of combination products falls under the Medical Device Regulation (MDR) and the In Vitro Diagnostic Regulation (IVDR), depending on the nature of the products. If neither component is authorized, the combination is treated as a new medical device or medicinal product. The European Medicines Agency (EMA) or national authorities will evaluate the submission, requiring comprehensive evidence of safety and performance. The classification of the combination (as a device or drug) will guide the regulatory pathway.
Health Canada regulates combination products under the Food and Drugs Act. If neither component is authorized, the product is treated as a new drug or medical device, requiring a new submission. Health Canada reviews the safety, efficacy and quality of the combination, similar to the FDA and EMA processes.
Q: Does the FDA have any guidance for combining non-approved products?
A: Yes, the FDA provides guidance on the regulation of combination products, which includes scenarios where non-approved products are involved. While there isn’t a specific guideline solely for combining non-approved products, the FDA’s framework for combination products outlines the regulatory pathways and considerations for products composed of multiple components.
While combining non-approved products presents unique challenges, the FDA’s framework provides a pathway for evaluation, emphasizing the need for comprehensive evidence of safety and efficacy. Engaging with the FDA early in the development process is advisable to navigate the regulatory landscape effectively.
Q: In the case of novel-novel codeveloped, is it expected from FDA and EMA that the contribution of the individual components is evaluated within the first trial proposed for codevelopment or is it acceptable to demonstrate this following the first codevelopment proof of concept study?
A: In the context of novel-novel codeveloped products, both the FDA and EMA have specific expectations regarding the evaluation of contributions from individual components.
- FDA perspective:
Initial trials: The FDA generally expects that the first trial proposed for codevelopment should assess the combined product’s safety and efficacy. However, it is also recognized that separating the contributions of individual components may not always be feasible in early-stage trials, especially if the primary goal is to establish proof of concept.
Subsequent studies: It is acceptable to demonstrate the contributions of individual components in subsequent studies. The FDA encourages a flexible approach and sponsors can plan for further studies to differentiate the effects of each component after establishing initial proof of concept. - EMA Perspective:
First trial considerations: The EMA expects that the first proof of concept trial should also consider the interactions and contributions of each component. However, similar to the FDA, the EMA acknowledges that full evaluation might not be possible in early trials.
Separate assessments: The EMA typically prefers that any subsequent studies designed to evaluate the individual contributions of components are clearly outlined in the development plan. The regulatory authorities expect that the overall development strategy will eventually address the individual contributions to ensure a comprehensive understanding of the product’s profile.
While both FDA and EMA allow for flexibility in demonstrating the contributions of individual components, they prefer that initial trials at least begin to address these contributions, especially in terms of safety and efficacy. A clear development plan that outlines how these contributions will be evaluated over the course of the development process is important for both regulatory bodies. Engaging with regulatory authorities through pre-submission meetings can help clarify expectations for specific codevelopment scenarios.
Q: In the case of novel-novel codevelopment with 2 biologics, is it expected from regulators that dose ranging of the individual drugs is completed before codevelopment is proposed?
A: In the case of novel-novel codevelopment involving two biologics, regulatory expectations regarding dose-ranging studies can vary between the FDA and EMA, but there are some commonalities.
- FDA perspective:
Dose-ranging studies: The FDA generally expects that dose-ranging studies for each individual biologic may be conducted prior to initiating codevelopment. This is particularly important to establish a safe and effective dose range for each component, which can inform the design of combination studies.
Proposed codevelopment: While it is advantageous to have dose-ranging data before proposing codevelopment, the FDA can be flexible. If the combination product’s rationale supports the concurrent evaluation of both biologics, sponsors may propose a codevelopment plan that includes initial trials assessing the combination based on preliminary dosing data.
Engaging with the FDA early in the development process can clarify how much dose-ranging data is necessary before entering codevelopment.
- EMA perspective:
Dose-finding studies: The EMA also typically expects that dose-ranging studies for each biologic are conducted prior to codevelopment, particularly to ensure patient safety and to optimize dosing strategies.
Combination trials: If the biologics have established safety profiles and the rationale for their combination is strong, the EMA may allow for simultaneous codevelopment, but this would ideally be preceded by sufficient data on each component.
Development plan: A well-defined development plan that outlines how dose-ranging will be integrated into the codevelopment process can be beneficial. The EMA encourages clear communication about the rationale for the proposed development strategy.
Both the FDA and EMA generally prefer that dose-ranging studies for individual biologics be completed before codevelopment is proposed. However, there may be flexibility depending on the specific context, safety considerations and the strength of the rationale for combination therapy. Engaging with regulators early in the process is advisable to ensure alignment on expectations and study designs.
Q: In the case of novel-novel codevelopment with 2 biologics, is it expected from Regulators that efficacy is demonstrated for the individual drugs before codevelopment is proposed?
A: In the context of novel-novel codevelopment involving two biologics, regulatory expectations regarding the demonstration of efficacy for the individual drugs before proposing codevelopment can vary, but there are common themes between the FDA and EMA.
FDA Perspective:
- Efficacy evidence: The FDA generally expects that there should be some evidence of efficacy for each individual biologic before proposing codevelopment. This is particularly important to ensure that each component has a demonstrated therapeutic benefit, which can support the rationale for their combination.
- Proposed codevelopment: While it is ideal to have established efficacy for both components, the FDA may allow for codevelopment if there is a strong scientific rationale or if the individual biologics have good safety data and a clear mechanism of action suggesting potential additive or synergistic effects when combined.
- Engagement with the FDA: Sponsors are encouraged to have discussions with the FDA during pre-IND meetings to clarify expectations regarding efficacy data and how it relates to the codevelopment plan.
EMA Perspective:
- Demonstration of efficacy: The EMA also typically expects that efficacy for each individual biologic should be established prior to codevelopment. This helps ensure that both components have a validated therapeutic effect, which is crucial for justifying their combination.
- Combination studies: If the individual biologics show promise in terms of safety and preliminary efficacy, it may be feasible to propose codevelopment, particularly if the combination is supported by compelling scientific rationale.
- Communication with regulators: Similar to the FDA, engaging with the EMA early in the development process is advisable to understand specific data requirements and to align on the proposed development strategy.
Both the FDA and EMA generally expect that efficacy for the individual biologics be demonstrated before codevelopment is proposed. However, there may be some flexibility depending on the context, especially if there is a strong scientific rationale for the combination. Early engagement (an area of expertise for Clarivate Regulatory Consulting) with regulatory authorities can help clarify expectations for specific codevelopment scenarios.
Q: Regarding FDA, is the take-away that each component needs to be approved PRIOR to the combination being approved ?
A: In general, the FDA does not require that each individual component of a combination product be fully approved prior to the approval of the combination itself. However, there are some important nuances to consider:
- Independent approval not required: It is possible for the FDA to approve a combination product even if the individual components have not been previously approved. The approval process for the combination will evaluate the safety and efficacy of the product as a whole.
- Efficacy and safety evidence: While prior approval of individual components is not necessary, the FDA expects that sufficient evidence of safety and efficacy for the combination product is provided. This may include data from clinical trials specifically designed for the combination.
- Regulatory pathway: The regulatory pathway for the combination product will depend on the primary mode of action. If the combination is intended to be used together, the FDA will assess it as a unique entity and all relevant data regarding the components and their interactions will be considered.
- Pre-submission engagement: Engaging with the FDA through pre-submission meetings can help clarify expectations and requirements for the specific combination product, especially regarding the evidence needed for approval.
In summary, while the FDA does not require prior individual approvals for components of a combination product, it is critical that the combination itself demonstrates adequate safety and efficacy through rigorous testing. Early dialogue with the FDA can help navigate the regulatory landscape effectively.
Q: Are you aware of any precedent of registering a combination of two NCEs?
A: Yes, there are precedents for registering combinations of two new chemical entities (NCEs) in both the FDA and EMA regulatory frameworks. Here are some notable examples:
Examples of combination products with NCEs:
- ATRIPLA: This is a combination of three antiretroviral drugs (efavirenz, emtricitabine and tenofovir) for the treatment of HIV. While it includes established drugs, it demonstrates the regulatory pathway for combining multiple NCEs, as the individual components were not initially combined in the same formulation.
- Combining anticancer agents: Several combinations of novel anticancer agents, such as immune checkpoint inhibitors, have been developed and registered as combination therapies. For instance, the combination of pembrolizumab (KEYTRUDA) and lenvatinib (LENVIMA) for certain cancers shows how NCEs can be combined for therapeutic purposes.
- Dulaglutide and liraglutide: In diabetes management, various combinations of GLP-1 receptor agonists have been explored, even though they may not have been marketed as a single product initially. The development of combinations like these often involves the registration of new formulations with distinct mechanisms of action.
Regulatory considerations:
- Clinical trials: For combinations of NCEs, robust clinical trials are necessary to demonstrate the safety and efficacy of the combination as a single entity.
- Scientific rationale: A strong scientific rationale for the combination is crucial, including evidence of potential synergy or enhanced efficacy.
- Regulatory pathways: Each NCE may need to undergo its own regulatory evaluation unless they are being evaluated as a single combination product.
While there are examples of registering combinations of two NCEs, each case is evaluated on its own merits based on the evidence provided. Regulatory agencies like the FDA and EMA carefully consider the safety, efficacy and scientific rationale for such combinations. Engaging with regulatory authorities during development can help navigate these complex pathways.
Q: Can one develop combination without each of the components to be approved as monotherapy first?
A: In drug development, a combination therapy typically requires that each component of the combination has been tested and approved as a monotherapy, especially when it comes to regulatory approvals. This is because regulatory agencies, like the FDA or EMA, often need to evaluate the safety and efficacy of each individual drug before considering their use in combination.
However, there are exceptions where a combination can be developed without prior approval of each component as a monotherapy. For instance, if there’s strong scientific evidence or clinical rationale supporting the combination’s potential benefits, developers might pursue a combination approval directly. This approach often requires robust data demonstrating that the combination is safe and effective together.
Ultimately, the specific regulatory pathway can vary based on the types of drugs involved, their mechanisms of action, existing clinical data and the intended patient population. It’s essential to consult regulatory guidelines and possibly engage with regulatory agencies early in the development process to understand the requirements for a specific combination therapy.
Q: In the phase 2b study do you have comparison with monoarms?
A: In a phase 2b study, it is common to include comparison groups, such as monotherapy arms, to assess the efficacy and safety of the investigational treatment. Including monotherapy arms allows researchers to compare the combination treatment’s effects against the effects of each component when administered alone.
This design helps to determine whether the combination has additive or synergistic effects and can provide insights into the overall benefit-risk profile of the treatment. However, the specific design of the study, including the presence of monotherapy arms, will depend on the objectives of the trial and the nature of the therapies being evaluated.
If you have a specific study in mind, the study protocol or published results would offer the most accurate details regarding the inclusion of monoarms and the comparison methodology used.
Q: How do the organizations go about identifying the molecules and finding what would be the market for such combination?
A: Computational approaches are crucial for predicting effective drug combinations, as they enable the integration and analysis of vast amounts of biological data. The most popular methods nowadays are either based on systems biology, machine learning or a combination of both. Systems biology methods are based on the analysis of biological networks and require extensive biological knowledge. Machine learning models try to learn complex patterns from the data but are limited by the amount of training data. The choice of the method depends on the amount of data available in the therapeutic area of interest.
The discovery and translational science consulting team at Clarivate has experience implementing such methods. Some clients come with an indication in mind and we can proceed in 2 steps:
- Prioritize targets for indications, using a machine learning pipeline that leverages information from a curated database of molecular interactions (Metabase) and Cortellis Drug Discovery Intelligence;
- Identify drugs for those target and use a systems biology method to predict the effect of those combinations.
In other cases, the client has one drug of interest and computational methods (either machine learning or systems biology) are used to find the optimal partner for this drug.
Finally, for identifying what would be the market for such combination, real word data can be of great utility. Epidemiological data can be used to assess the size of the market. In addition, information about patented drugs, market trends, drug development stages, clinical trials discontinued due to toxicity and more can be leveraged using Clarivate tools for market assessment such as Cortellis Competitive Intelligence or Disease Landscape and Forecast. Clarivate Commercial Strategy Consulting services experts can assist you in that task.
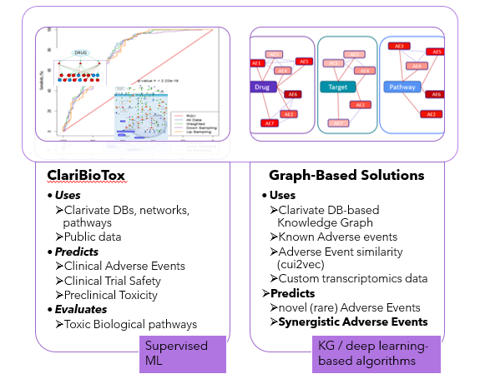
Q: You mentioned that safety for a combination can be predicted based on preclinical models. How predictive is that of safety in humans as that is very complex for a combination?
A: You are correct that predicting the safety of a drug combination in humans based solely on preclinical models is challenging. This is especially true if the preclinical model does not accurately represent the human disease (e.g., using cell lines versus 3D organoids).
In these situations, in which the relevant experimental data is lacking, we prefer using experimental data-agnostic prediction methods that leverage existing knowledge about drugs and diseases. In our Discovery and Translational science consulting team, we typically employ two main approaches: a supervised machine learning approach and a graph-based approach:
Machine learning-based approach:
This approach aims to predict toxicity and is flexible so it could be set up using or not using any experimental preclinical and clinical information. It can be applied in two scenarios:
- When we have experimental data for a drug or drug combination that is representative of the disease (good preclinical model, clinical data, etc.).
- When we use biological information about disease biomarkers, pathways, PPIs, etc (e.g., from CDDI, Metabase) and chemical properties of drugs as input.
Graph-based approach:
- This method utilizes direct and predicts new toxicity annotations for drug combinations from the OFF-X database (Clarivate’s safety database) and it does not require any experimental input data. These methods have good predictive power (AUC > 0.8). However, it requires that the drugs on the combo have at least some existing annotations, so it may not be suitable for entirely new drugs (not usually the case for drug combinations).
View the full webinar here. To learn more about how Clarivate discovery and preclinical solutions help innovators overcome complex scientific challenges, read our case study on the use of predictive analytics to identify promising drug combinations here.
DISCLAIMER: Clarivate Consultants responded to the questions asked based on their experience and knowledge, however, such responses are not intended to provide legal advice or the provision of legal services. Clarivate Consultant’s responses are not, and shall not be construed as, a substitute for legal advice from a qualified legal services provider validly licenced to provide such advice. Clarivate does not warrant or guarantee any decision to act (or not to act) upon the response’s provided or information contained in the original webinar or this blog.




Related insights
The latest news, technologies, and resources from our team.





